LESSA: a Library of Building Blocks for Efficient, Scalable and Service-oriented Algorithms
1.Machine Learning Foundation Techniques and Applications
We are dedicated to developing foundation techinques in machine learning, including but not limited to foundation models (e.g. large language/graph models), gradient desecent optimizers, distrbuted learning systems, deep learning compilers and automatic machine learning (AutoML), and further develop practical applications to solve real-world challenges
Example work:
|
2. Compact Computing for Big Data
As data volume increases exponentially and data compression is popularly used in data centers, I would expect that directly operating on compressed data would become commonplace in the future. However, parallel processing of compressed data is a challenging proposition for both shared-memory and distributed-memory systems and the challenges could come from constrained random accesses to uncompressed content, independent decompression of data blocks, balanced distribution of data, memory and computation, adaption of existing algorithms and applications to meet the requirements of compressed data processing and so on. Based on these concerns, I am conceiving of a new concept of computing and name it as Compact Computing tentatively. In general, Compact Computing targets robust, flexible and reproducible parallel processing of big data and consists of three core components, in principle: (1) tightly-coupled architectures, (2) compressive and elastic data representation, and (3) efficient, scalable and service-oriented algorithms and applications. In this context, component 1 can comprise conventional CPUs, a diversity of accelerators (e.g. FPGAs, GPUs, MIC processors and etc.) and fast interconnect communication facilities; component 2 concentrates on data structures and formats that enable efficient on-the-fly streaming compression and decompression; and component 3 targets the development of algorithms and applications that enable robust and efficient processing of big data streams in parallel. By centering around data, Compact Computing enables full-stack computation by tightly coupling algorithms with systems.
Example work:
|
3. Large-Scale Biological Sequence Analysis System
As part of my LESSA library, this project has been the core of my research on parallel and distributed algorithm design for bioinformatics, by employing a variety of tightly-coupled and loosely-coupled computing architectures, including heterogeneous computers with accelerators (e.g. Intel SSE, Intel AVX, Intel Xeon Phis, NVIDIA GPUs and AMD GPUs), cluster computing and cloud computing. My final objective is to establish an analysis system for large-scale biological sequences, in order to solve some critical and bottleneck problems in bioinformatics and computational biology, such as genome sequencing based on high-throughput sequencing technologies, meta-genomics, motif discovery, sequence alignment, and phylogenetic inference. Fig. 1 illustrates the diagram of an imaginaory biological data analysis system.
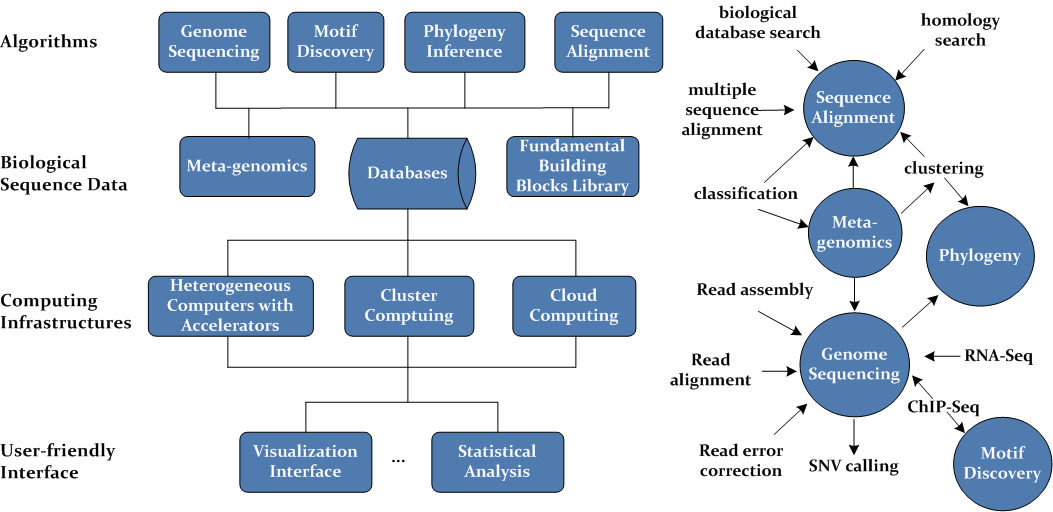
|
| Fig. 1 System diagram for large-scale biological sequence analysis |
Example work:
(The full lists of my software and publications are available here)- Liu Y, Maskell DL, Schmidt B: "CUDASW++: optimizing Smith-Waterman sequence database searches for CUDA-enabled graphics processing units". BMC Research Notes, 2009, 2:73
- Liu Y, Schmidt B, Maskell DL: "MSA-CUDA: multiple sequence alignment on graphics processing units with CUDA". 20th IEEE International Conference on Application-specific Systems, Architectures and Processors (ASAP 2009), 2009, 121-128 (Best Paper Award)
- Liu Y, Schmidt B, Maskell DL: "MSAProbs: multiple sequence alignment based on pair hidden Markov models and partition function posterior probabilities". Bioinformatics, 2010, 26(16): 1958 -1964
- Liu Y, Schmidt B, Liu W, Maskell DL: "CUDA-MEME: accelerating motif discovery in biological sequences using CUDA-enabled graphics processing units". Pattern Recognition Letters, 2010, 31(14): 2170 - 2177
- Liu Y, Schmidt B, Douglas L. Maskell: "CUDASW++2.0: enhanced Smith-Waterman protein database search on CUDA-enabled GPUs based on SIMT and virtualized SIMD abstractions". BMC Research Notes, 2010, 3:93
- Liu Y, Schmidt B, Maskell DL: "Parallelized short read assembly of large genomes using de Bruijn graphs". BMC Bioinformatics, 2011, 12:354
- Kuttippurathu L, Hsing M, Liu Y, Schmidt B, Maskell DL, Lee K, He A, Pu WT, and Kong SW: "CompleteMOTIFs: DNA motif discovery platform for transcription factor binding experiments". Bioinformatics, 2011, 27(5): 715-717
- Liu Y, Schmidt B, Maskell DL: "CUSHAW: a CUDA compatible short read aligner to large genomes based on the Burrows-Wheeler transform". Bioinformatics, 2012, 28(14): 1830-1837
- Liu Y and Schmidt B: "Long read alignment based on maximal exact match seeds". Bioinformatics, 2012, 28(18): i318-i324 (also from ECCB 2012)
- Liu Y, Schroeder J, Schmidt B: "Musket: a multistage k-mer spectrum based error corrector for Illumina sequence data". Bioinformatics , 2013, 29(3): 308-315
- Liu Y, Wirawan A, Schmidt B: "CUDASW++ 3.0: accelerating Smith-Waterman protein database search by coupling CPU and GPU SIMD instructions". BMC Bioinformatics, 2013, 14:117.
- Liu Y, Schmidt B: "CUSHAW2-GPU: empowering faster gapped short-read alignment using GPU computing". IEEE Design & Test, 2014, 31(1): 31-39
- Ripp F, Krombholz CF, Liu Y, Weber M, Schaefer A, Schmidt B, Koeppel R, Hankeln T: "All-Food-Seq (AFS): a quantifiable screen for species in biological samples by deep DNA sequencing". BMC Genomics, 2014, 15:639
- Liu Y, Tran TT, Lauenroth F, Schmidt B: "SWAPHI-LS: Smith-Waterman algorithm on Xeon Phi coprocessors for long DNA sequences". 2014 IEEE International Conference on Cluster Computing (Cluster 2014), 2014, pp. 257-265 (Best Paper Award Recommendation)
- Liu Y, Schmidt B: "SWAPHI: Smith-Waterman protein database search on Xeon Phi coprocessors". 25th IEEE International Conference on Application-specific Systems, Architectures and Processors (ASAP 2014), 2014, pp. 184-185
- Liu Y, Loewer M, Aluru S, Schmidt B: "SNVSniffer: an integrated caller for germline and somatic SNVs based on Bayesian models". 2015 IEEE International Conference on Bioinformatics and Biomedicine (BIBM15), 2015, pp. 83-90.
- Liu Y, Loewer M, Aluru S, Schmidt B: "SNVSniffer: an integrated caller for germline and somatic single-nucleotide and indel mutations". BMC Systems Biology, 2016, 10(Suppl 2):47
- Pan T, Flick P, Jain C, Liu Y and Aluru S: "Kmerind: A flexible parallel library for k-mer indexing of biological sequences on distributed memory systems". 7th ACM Conference on Bioinformatics, Computational Biology, and Health Informatics (ACM-BCB 2016), 2016, pp. 422-433
- Chan Y, Xu K, Lan H, Liu W, Liu Y and Schmidt B: ”PUNAS: a parallel ungapped-alignment-featured seed verification for next-generation sequencing read alignment”. 31st IEEE International Parallel and Distributed Processing Symposium (IPDPS 2017), 2017, pp. 52-61.
- Lan H, Liu W, Liu Y and Schmidt B: ”SWhybrid: a hybrid parallel framework for large-scale protein sequence database search”. 31st IEEE International Parallel and Distributed Processing Symposium (IPDPS 2017), 2017, pp. 42-51.